Cystisk fibros är en medfödd metabolisk sjukdom. Kroppsvätskor som saliv, bronkialslem eller bukspottkörtelnutsöndringar är mycket tuffare än vanligt på grund av genetisk predisposition. Konsekvenserna inkluderar andningsproblem och matsmältningsbesvär. Cystisk fibros är inte härdbar. Med konsekvent behandling kan emellertid sjukdomsförloppet bromsas. Läs här vilka symtom som orsakar cystisk fibros och hur du behandlar den.

Cystisk fibros: kort översikt
- Beskrivning: ärftlig ämnesomsättning som orsakar tuff slembildning i lungorna och andra organ
- symptom: Andningsproblem, irriterande hosta, lunginfektion, misslyckande med att frodas, matsmältningsbesvär, svår diarré, fet lever, minskad fertilitet
- orsakar: Arv av defekta gener som påverkar konsistensen av kroppsvätskor, utbrott av sjukdomen endast om båda föräldrarna ärver en sjuk gen (dominerande recessiv arv)
- diagnos: Blodtest för immunreaktivt trypsin (IRT), pankreatitassocierat protein (PAP), svettest, genetiskt test
- behandling: mukolytiska medel, bronkodilaterande medel, inandning, antibiotika för infektioner, kortison, CFTR-modulatorer, lungtransplantation
- prognos: inte härdbar, naturligtvis beroende av svårighetsgraden och tidpunkten för diagnosen, förkortad livslängd
Cystisk fibros: beskrivning
Cystisk fibros (även kallad cystisk fibros) är en ärftlig metabolisk sjukdom. Bildningen av olika kroppsvätskor störs. Utsöndringarna av lungorna, bukspottkörteln och andra organ är mer viskösa än hos friska människor.
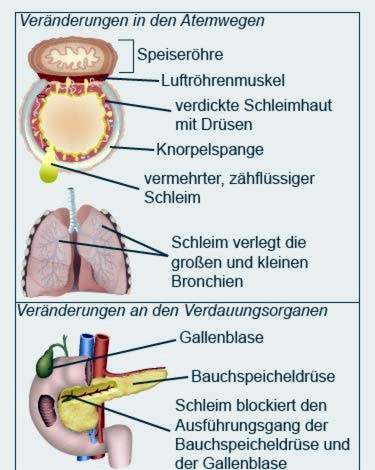
Tufft slem
Det tuffa slemet täpper bland annat de små grenarna i bronkierna och kanalerna i de inre organen. Andning och matsmältning påverkas särskilt. Under sjukdomen kan organen fungera sämre och sämre.
Fel i det genetiska materialet
Orsaken till sjukdomen är defekter i det genetiska materialet. Cystisk fibros är därför inte härdbar. Tid för diagnos och svårighetsgrad av symtom kan variera mycket individuellt. Hos många barn gör cystisk fibros en massiv inverkan från födseln, i andra fall erkänns det senare.
Cystisk fibros: symtom
Cystisk fibros-symtom kan variera mycket från patient till patient. Sjukdomen påverkar funktionen hos olika organ, men särskilt lungorna och matsmältningssystemet.
Känn igen tidiga tecken
De första symtomen på cystisk fibros varierar individuellt. I de flesta fall visas symtom på cystisk fibros inom det första leveåret. Således kan sjukdomen vanligtvis diagnostiseras tidigt och starta snabbt med en terapi. Vissa patienter har dock betydande klagomål endast i tonåren. Inte varje person som berörs visar hela utbudet av möjliga symtom. Svårighetsgraden av symtomen varierar också.
Förändrade kroppsvätskor
Vid cystisk fibros störs bildningen av de så kallade kloridjonkanalerna i cellerna. Detta ändrar sammansättningen av kroppsvätskor. Det enklaste sättet att upptäcka denna förändring i svett hos de drabbade. Din svett är saltare än friska människor. Salterna natrium och klorid, som tillhör de så kallade elektrolyterna, berikas i deras svett. Som ett resultat av svettning förlorar patienter som lider av cystisk fibros alltmer kroppssalter.
Olika symtom på cystisk fibros
Sjukdomen drabbar en rad olika organsystem. Ofta visas de första symtomen på cystisk fibros i lungorna och matsmältningskanalen. Under livet kan ytterligare klagomål läggas till. Genom riktad terapi kan symtomen behandlas väl. Emellertid kan symtomen också nå hotfulla proportioner. Det är särskilt farligt när bronkierna tränger igenom det viskösa slemet. Då kan patienterna kvävas i extrema fall.
Cystisk fibros: symtom på lungan
Andningsproblem och irriterande hosta
I de flesta fall förekommer symtom i lungorna i cystisk fibros endast hos något äldre spädbarn. Nyfödda har vanligtvis inga andningsproblem. Cystisk fibros-symtom uttrycks ofta i form av en hosta-halsliknande, kronisk, irriterande hosta hos något äldre barn. Slemet i deras andningsorgan är ökat, tufft och visköst. Detta hindrar luftflödet i lungorna. Med tiden utvecklas en progressiv andningsbesvär.
Ofta infektioner
Ökad slemproduktion i lungorna gör det lättare för bakterier att kolonisera och orsaka infektion. Återkommande lunginflammation eller bronkialinfektioner orsakas huvudsakligen av bakterier som stafylokocker och Pseudomonas. Den störda saltbalansen i lungorna hindrar också kroppens försvar. Dessutom kan lungblödningar uppstå. Ett typiskt tecken på detta är hosta av blodblandat slem.
Även om lungorna är skadade från en tidig ålder, är de första symtomen på cystisk fibros i luftvägarna ofta först i grundskolealdern eller till och med senare. Symtomen förekommer ibland endast när stora delar av lungvävnaden redan har förstörts eller luftvägarna är hårt förträngda.
Cystisk fibros: symptom på bukspottkörteln
Hos patienter med cystisk fibros blir bukspottkörteln ofta inflammerad. Den utsöndrar en sekretion som innehåller bland annat enzymer för matsmältning av fett och socker. Hos cystisk fibros drabbas utsöndringen på grund av dess viskositet tillbaka och orsakar inflammation.
När processen fortskrider blir pankreasvävnaden härdad och ärr. Läkare talar om fibros. Fibrosen förstör gradvis bukspottkörteln. Förutom gallan bildar bukspottkörteln också insulin, vilket behövs bland annat för att använda sockret i kroppen. Patienter som är lite äldre (från tonåren) utvecklar ofta diabetes mellitus.
Cystisk fibros: galla symptom
Bukspottkörteln och gallblåsan delar en gemensam kanal i tarmen. Därför kan återflödet av bukspottkörtelnutsöndringar också orsaka inflammation i gallblåsan. Ofta bildas gallsten som helt kan blockera gallblåsan från gallblåsan.
Cystisk fibros: symtom på matsmältningskanalen
Förutom klagomål på lungorna påverkar symtom på cystisk fibros främst matsmältningen. På grund av bristen på galla, till exempel, försämras fettutsluten. Ofta tolererar patienter fet mat inte. Den intagna maten utsöndras till stor del osmält igen. Typiska är då mycket voluminösa och mjuka tarmrörelser.
Diarré och tillväxtproblem
Påverkade barn, inklusive spädbarn, lider ofta av svår diarré. Även om de dricker och äter bra ökar de knappt. Tillväxtstörningar och undernäring är därför ytterligare klassiska konsekvenser av sjukdomen.
Sådana klagomål inom området för matsmältningen kan emellertid också förekomma vid andra sjukdomar. Endast i kombination med andningsproblem är de därför en karakteristisk indikation på cystisk fibros, vilket definitivt bör undersökas.
anal prolaps
I den fortsatta kursen kan cystisk fibros orsaka olika komplikationer i matsmältningskanalen. Det vanligaste är en så kallad anal prolaps. Vid anal prolaps buknar tarmslemhinnan ut från anus. En sådan incident måste behandlas kirurgiskt så snart som möjligt.
tarmobstruktion
Involvering av tarmen (invagination) eller tarmhinder (ileus) är också vanligt. Båda komplikationerna är förknippade med svår magsmärta och betydande matsmältningsproblem. Smärtan uppstår vanligtvis i spurts, särskilt efter att ha ätit. En tarmhinder är dödlig om den inte behandlas. Spasmodisk, akut buksmärta bör därför alltid klargöras av en läkare.
Cystisk fibros: symtom i levern
fettlever
Under bakflödet av gallan lider också levern. Hos många patienter utvecklas fet lever under sjukdomsförloppet. Trötthet, aptitlöshet, uppblåsthet och flatulens och i sällsynta fall känslor av tryck eller smärta i övre buken.
levercirros
I sällsynta fall utvecklas en krympande lever (levercirrhos) där levern störs kraftigt i sin funktion. Det manifesteras först i form av gulsot (gulsot). Tecken på gulsot är den gulaktiga missfärgningen av vitt i ögonen. Efter lång tid uppstår också hjärtproblem och prestandan hos de drabbade fortsätter att minska.
Cystisk fibros: minskad fertilitet
Mer än hälften av alla manliga patienter är infertila. Även om de i de flesta fall kan bilda bördigt spermier, kan de inte komma igenom vas deferens eftersom de är blockerade av visköst slem.
Berörda kvinnor är vanligtvis mindre bördiga. I allmänhet kan de ta emot och leverera ett barn. Men i deras äggledare samlas tufft slem, vilket spermierna knappast kan tränga igenom. Speciellt vid en högre ålder minskar sannolikheten för graviditet snabbt.
Cystisk fibros: symtom hos barn
Cystisk fibros är en genetisk sjukdom. Det har alltid funnits sedan födseln. Men de klassiska symptomen på cystisk fibros förekommer inte alltid i barndomen. Men det finns ofta redan ospecifika instruktioner som bör följas. Detta gäller särskilt om fall av cystisk fibros redan har inträffat i familjen.
Uppsvälld buk
En indikation på att det finns en ämnesomsättning är till exempel en uppblåst buk under lång tid. Ofta lider barnen av diarré. Hos nyfödda kan en markant försenad första avföring (Kindspech) vara en indikation på cystisk fibros. I många fall förekommer tillväxt- och tillväxtstörningar även om barnen äter med sug. Endast i sällsynta fall leder det också till förstoppning (förstoppning) till följd av sjukdomen.
Skramlande andning
Andra symtom som kan indikera cystisk fibros inkluderar skrattande andning och allvarlig rastlöshet. Många barn lider av kronisk inflammation i bihålorna. Dessa märks främst av inte exakt lokaliserbar smärta i ansiktet. Nasala polypper är vanligare hos barn med cystisk fibros än hos friska barn.
Om barn lider av andningsproblem eller matsmältningsbesvär längre, bör en läkare alltid konsulteras som en försiktighetsåtgärd. Hos barn kan livshotande situationer snabbt uppstå eftersom de själva inte kan formulera sina klagomål eller deras svårighetsgrad inte kan bedöma.
Cystisk fibros: orsaker och riskfaktorer
Cystisk fibros orsakas av en genetisk defekt. Den patologiska förändringen ligger på den sjunde kromosomen i den så kallade CFTR-genen.
CFTR-genen (cystisk fibros transmembranregulatorgen) innehåller konstruktionshandboken för en kanal genom vilken kloridjoner kommer in i cellerna. De defekta kloridjonkanalerna blockerar transport av salt till vissa kroppsceller hos patienter med cystisk fibros.
De drabbade körtelcellerna men istället för den annars flytande utsöndringen tufft slem från. I lungorna, paranasala bihålorna, bukspottkörteln, tarmen, i gallvägarna och i gonaderna bildas viskösa slemutsöndringar med högt saltinnehåll.
Cystisk fibros: hur hotat är mitt barn?
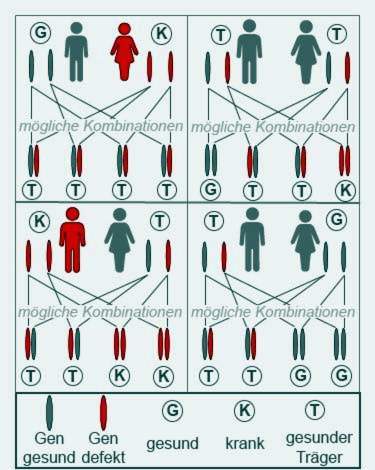
Cystisk fibros bryter bara ut när båda föräldrarna överför en patologiskt förändrad gen till sitt barn. Dessa föräldrar är då främst båda själva friska, men bärare av genen.
Personer med cystisk fibros är endast delvis bördiga. Vissa patienter blir fortfarande föräldrar. Sjuka fäder eller mödrar överför emellertid alltid en sjuk gen, eftersom båda CFTR-generna har cystisk fibrosinformation. Men deras barn blir bara sjuka om de också får en sjuk gen från den andra föräldern.
Par vars familjer redan har haft cystisk fibrosfall bör söka genetisk rådgivning innan de planerar en graviditet.
Preimplantatorisk genetisk diagnostik
Föräldrar som kan föda cystisk fibros kan genomgå genetisk diagnos för preimplantation. Vid genetisk testning av preimplantation befruktas först oocyterna artificiellt. De första celldelningarna sker i provröret (in vitro).
Innan ett embryo sätts in testas det för förändrade genegenskaper. Endast embryon implanteras sedan som inte bär den sjuka genen. Oavsett detta kan det också undersökas under graviditeten om barnet kommer att utveckla CF senare.
Cystisk fibros: undersökningar och diagnos
Till skillnad från för några år sedan genomgår de flesta sjukhus idag rutinmässigt neonatal screening. Detta inkluderar studier på cystisk fibros.
Screening i blod och svett
För screening tas blod från den nyfödda. Cystisk fibrostest innefattar flera steg:
blodtest
Test för förhöjda nivåer av immunreaktivt trypsin (IRT) och pankreatitassocierat protein (PAP). I händelse av avvikelser sker svettest.
svetstestet
Cystisk fibrospatienter har en betydligt högre salthalt hos svett än friska människor. För svettest av cystisk fibros mäts innehållet av salterna natrium och klorid i kroppssvetten. Hos barn samlas svett på underarmen och analyseras sedan. Om en misstank uppstår här kommer ett genetiskt test att genomföras.
genetiskt test
Hos patienter med cystisk fibros har den så kallade CFTR-genen, som tillhandahåller planen för specifika jonkanaler, förändrats. Denna konstruktionshandbok är lång, den består av cirka 6500 baspar. Överallt kan ett fel krypa till kod, men felen har olika effekter. Därför testas endast de vanligaste avvikelserna i koden.
Familjehistoria ger tips
Om ingen lämplig neonatal screening har utförts och den misstänkta cystiska fibrosen kommer upp senare är familjeläkaren eller internisten rätt person att kontakta. I en inledande konversation registrerar denna person sjukhistorien (anamnesis). Vid misstänkt cystisk fibros uppmärksammas familjehistoria särskilt.
Fysisk undersökning
Därefter sker en fysisk undersökning. Läkaren lyssnar på lungorna och skannar de inre organen. Han kan redan utesluta några andra tillstånd som är förknippade med symtom som liknar cystisk fibros.
Dessutom kan röntgenundersökningar visa andnings- och lungstopp. Laboratorieundersökningar ger indikationer på funktionella begränsningar av de inre organen. Även hos vuxna som misstänks för cystisk fibros ger ett svettest viktiga bevis för diagnosen.
Testa familjemedlemmar
Om cystisk fibros upptäcks i en familj är det vettigt att alla andra familjemedlemmar också genomgår en undersökning. Cystisk fibros kan också förekomma i försvagade former. Det tar ofta många fler år för cystisk fibros att manifestera sig med tydliga symtom. Ändå är tidig diagnos och cystisk fibrosterapi också viktigt för de drabbade för att öka livslängden.
Cystisk fibros: behandling
Cystisk fibros är inte härdbar. Barn födda med cystisk fibros drabbas av effekterna av sjukdomen under hela livet. Emellertid kan en kombination av fysioterapi, medicinering och inandning betydligt bromsa utvecklingen av sjukdomen. En cystisk fibrosterapi bör därför främst uppnå att de drabbade kan leva ett så normalt liv som möjligt.
Lära sig att leva med sjukdomen
Speciellt för barn är det viktigt att lära sig hantera sjukdomen på lång sikt. Barn med cystisk fibros bör läras så tidigt som möjligt vad sjukdomen betyder och hur den påverkar kroppen. Här kan en sjukhusvistelse med specialutbildningsenheter vara användbar. Under processen lär sig barn och föräldrar att mata sig själva, hur sport ser ut och hur de beter sig bäst i kritiska situationer.
Hjälp för lungorna
Det finns olika alternativ för att behandla symtomen. Beroende på patientens ålder och svårighetsgraden av symtomen rekommenderas olika metoder.
Mukolytiska medel
Cystisk fibrospatienter lider mest av lungproblem. Genom regelbunden inandning med speciella tillsatser (mukolytika) löses det viskösa slemet och kan lätt hostas av.
Bronchiala dilateringsmedel
Så kallade beta-2-sympatomimetika ökar också bronkierna, vilket dessutom underlättar andningen.
Antibiotika mot bakterier
På grund av dålig ventilation i lungorna lider människor med cystisk fibros av bakteriella infektioner i luftvägarna mycket oftare. Å andra sidan hjälper antibiotika som administreras i god tid. I vissa fall är det vettigt att andas in dessa permanent.
Antiinflammatoriska läkemedel
Hos många patienter är andningsorganen ofta eller kroniskt inflammerade. Då hjälper antiinflammatoriska läkemedel som kortison.
CFTR modulatorer
Samtidigt har de första läkemedlen utvecklats som förbättrar jonkanalernas nedsatta funktion. De arbetar emellertid bara för vissa mutationer och därmed bara för en liten del av patienterna. Deras effektivitet är också begränsad. Förbättringen av läkemedlen undersöks intensivt. För första gången skulle de börja med orsaken till sjukdomen snarare än bara med symtomen.
Lungetransplantation – det sista hoppet
I svåra fall är en lungtransplantation också ett alternativ. Så drastiskt som detta steg verkar till en början kan många patienter leva ett betydligt lägre belastat liv efteråt.
Mata ordentligt i cystisk fibros
Eftersom cystisk fibros stör också matsmältningen, måste patienterna noga uppmärksamma sin kost. Du bör föredra en diet med mycket protein och kolhydrater.
Det finns också vitamintillskott och mineraler. Det senare ersätter de salter som svettar patienterna i stora mängder. Eftersom bukspottkörteln inte fungerar ordentligt får barn matsmältningsenzymer som tillägg till måltider.
Upptäck vaccinationer
Vaccination är särskilt viktigt för patienter med CF. Med dem har bakterier enklare spel och de blir ofta svårare än patienter som inte är förbelastade. Speciellt rekommenderas vacciner mot mässling och pneumokocker. Dessutom bör varje år vara ett influensavaccin.
Cystisk fibros: sjukdomsförlopp och prognos
Cystisk fibros orsakas av förändring i genomet och är därför inte härdbart. För personer med cystisk fibros reduceras livslängden och livskvaliteten vanligtvis avsevärt. Utan behandling förvärras hälsotillståndet snabbt och de drabbade lever ofta inte länge.
Med snabb och konsekvent behandling kan sjukdomsförloppet bromsas avsevärt. Under tiden lever patienterna mycket längre än för några år sedan. Den genomsnittliga livslängden för cystisk fibros är för närvarande cirka 40 år. Men många lever också med sjukdomen i 50 år och mer.
Komplikationer och följare
Även vid intensiv behandling kan komplikationer uppstå gång på gång vid cystisk fibros. Oftast finns det akut andningsbesvär på grund av dålig lungventilation. Enskilda lungor kan till och med kollapsa (alektas).
Ofta utvecklas kronisk bronkit eller lunginflammation. Svamp kan också påverka lungorna.
Dessutom kan förskjutningar i vätske- och elektrolytbalansen utlösa chock och cirkulationsfel.
Andra komplikationer och följder inkluderar:
- kronisk leversjukdom, särskilt cirros
- Gallblåsainflammationer och gallsten
- kronisk inflammation i bukspottkörteln
- störd hjärtfunktion
- akut tarmobstruktion (ileus)
- Intestinal invagination
- undernäring
- Diabetes mellitus
- Begränsad fertilitet hos kvinnor eller infertilitet hos manliga patienter
Cystisk fibros är en ärftlig sjukdom, förebyggande är därför inte möjligt. Människor med risk för familjerisk bör söka genetisk rådgivning om de vill få barn. Ett genetiskt test utförs och det undersöks om CFTR-genen förändras. Beroende på om en eller båda parterna bär genen kan risken för avkomman beräknas.
Under tiden är genetisk diagnos (PID) vid cystisk fibros förimplantation också möjlig i Tyskland. Förutsättningen för detta är alltid godkännande av en etisk kommitté. Oocyter befruktas utanför livmodern och endast embryon utan problematiska cystiska fibroser används.
Mer information
riktlinjer:
- S2 Consensus Guideline ”Diagnosis of Cystic Fibrosis” från Society for Pediatric Pulmonology (2013)
- S3-riktlinje ”lungsjukdom vid cystisk fibros” av Society for Pediatric Pulmonology (2013)
Stödgrupper:
- Cystisk fibros e.V.